应用串联质谱的数据搜索蛋白质数据库,已经发展成为成熟的研究生物样本中蛋白质组成的方法。然而,如果被搜索的参考蛋白质数据库只是包含了已知的蛋白质,那么利用该方法发掘新蛋白或修饰蛋白的能力就有一定的局限。
离子阱质谱仪,由于其具有高灵敏性和快速的扫描速度可以确保蛋白质组的高覆盖率,在蛋白质基因组学研究中使用最为广泛。然而,应用离子阱质谱仪得到的数据解析度和准确性范围在0.2~0.5 Da (200~500 ppm在1000 m/z),存在一定的局限性。较低的灵敏度使离子阱质谱仪需要高的质量允差度(mass tolerance),因此增加了搜索空间并降低了搜索的准确度。
高灵敏度的质谱仪器,例如FT ICR和Q-TOF都具有很好的解析度,可以达到亚-ppm级的质量灵敏度,这将可以减低搜索空间并增加蛋白质数据库搜索时的准确性。然而,高灵敏度的质谱仪存在一个缺点就是相对较低的数据获取速度,导致在分析复杂肽段混合物时的采样不足,并进而影响蛋白质组的覆盖率。
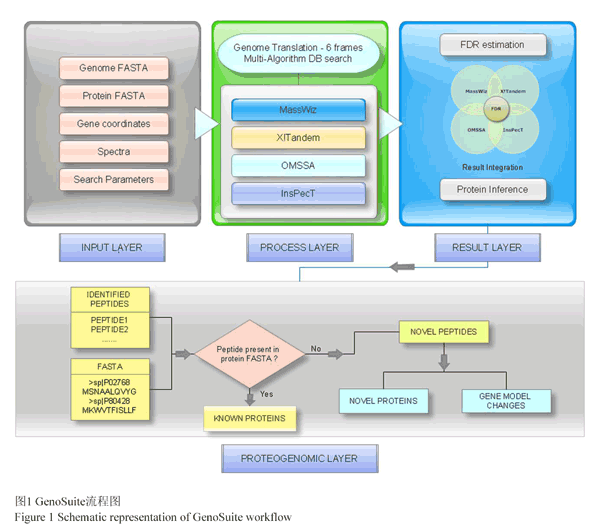
Bacterial Proteogenomic Pipeline是基于Java单机运行具有图形化界面的跨平台细菌蛋白质基因组学分析工具。该细菌蛋白质基因组学分析工具供包含六个可以使用命令行或Java Swing图形化界面运行的模块:Parse Protein Information模块将读取一个包含基因组读码框位置信息的FASTA格式的蛋白质库和一个包含已注释基因或蛋白质的所有信息的TSV/CSV文件,创建一个已知蛋白质的GFF3文件;Compare And Combin可选模块使用另外一个FASTA数据库作为参考选项,进一步对Parse Protein Information模块创建的GFF文档和对应的FASTA文档添加信息;Genome Parser模块依据细菌基因组序列创建六码框蛋白质数据库;Create Decoy DB可选模块用来创建诱饵数据库;Combine Identifications模块将外部搜索引擎以mzTab文档格式输入,对鉴定的PSMs进行验证和FDR过滤;Analysis模块可以对鉴定的肽段进行分析,并可视化每个肽段对应的不同的鉴定的PSMs的数目。Bacterial Proteogenomic Pipeline支持将任何鉴定搜索算法和后处理算法得到的肽段鉴定转换为mzTab格式输入,可以对不同实验条件下鉴定得到的肽段可视化和比较分析。并且所有的蛋白质和肽段信息都可以输出到GFF3格式文档中,可以利用自身模块实现可视化检验,也便于在常用的基因组阅览器上近一步分析验证。
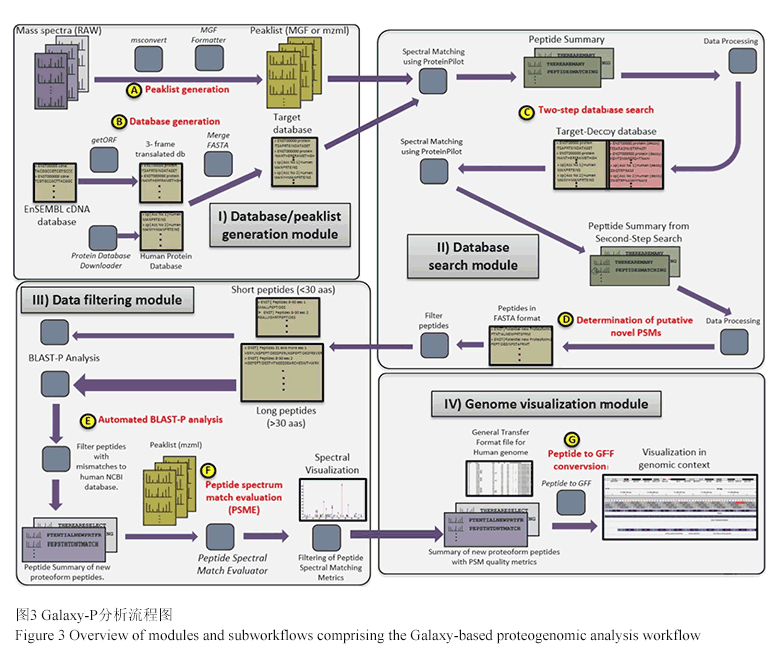
Galaxy-P分析的灵活性的体现之一就是数据库构建的灵活性,使得Galaxy-P可以针对不同的样品进行不同的数据库改变。这种灵活性带来便利和针对性在研究者使用该分析流程对人类唾液的蛋白质基因组学的演示分析中得到体现。研究人员发现在PSMs搜索分析中使用人类蛋白质数据库加入人类口腔微生物的蛋白质数据库组成综合数据库,比单独使用人类蛋白质数据库要多发现两倍的新序列变异体。导致这一问题出现的原因就是:微生物序列的缺失会导致串联质谱得到的来自非宿主的肽被迫和宿主的蛋白质相匹配,增加了假阳性并迫使PSMs为得到可接受的FDR必须有一个更高的得分值,因此就降低了新肽序列可信匹配的数量。这一发现说明在进行蛋白质基因组学的研究中,在样品包含非宿主的蛋白质情况下进行的蛋白质基因组学分析,如果仅仅使用宿主的蛋白质序列进行数据库搜索,其他结果会受到很大影响。
蛋白质基因组学分析往往依靠单个的PSMs来确定潜在的新蛋白序列,考虑到单个PSMs带来的潜在的假阳性,Galaxy-P的蛋白质基因组学分析流程中提供了多中水平的质量控制和过滤。除了在数据库搜索模块使用多个数据搜索引擎来改善结果的可信度外,在关键的第三模块中BLASTP方法的使用和自行开发的PSME (peptide spectrum match evaluation)工具是PSMs质量控制的重要一环。BLASTP中和NCBI数据库的比对可以进一步过滤掉和已知序列匹配的PSMs。PSME 不仅提供了一种可视化串联质谱图谱及其对应假定序列匹配的工具,还可以用户自行设定多种PSM质量相关的参数来对质谱进行过滤。在Galaxy-P 演示数据的分析中BLASTP的分析将9333个PSMs减少点1630个,PSME高严谨度PSM质量标准的限定更是将新肽序列匹配的数量减少到55个。
Galaxy-P的蛋白质基因组学分析流程的最后一步是通过自开发的“Peptides to GFF”工具来将肽段的氨基酸序列转换为IGV (integrated genome viewer)兼容的格式,从而实现在基因组中的可视化和阐释。新肽片段在基因组上的可视化有利于进一步的对这些肽段所对应假定新蛋白质可能性的评估分析。
虽然,基于Galaxy-P的蛋白质基因组学分析流程尽管包含大约140多个步骤,但整个流程在参数优化设定的情况下,只需要一次单击就可以完成整个流程,最要的是每个模块中的分流程都可以按需求单独自我运行。这些整个流程可以通过一个网络连接或一个保存的Galaxy流程文档而与其他人分享,和流程文档一样Galaxy-P的历史文档也可以被分享,历史文档中包含了重现整个分析流程所需要的所有的软件和各种输入与输出数据。
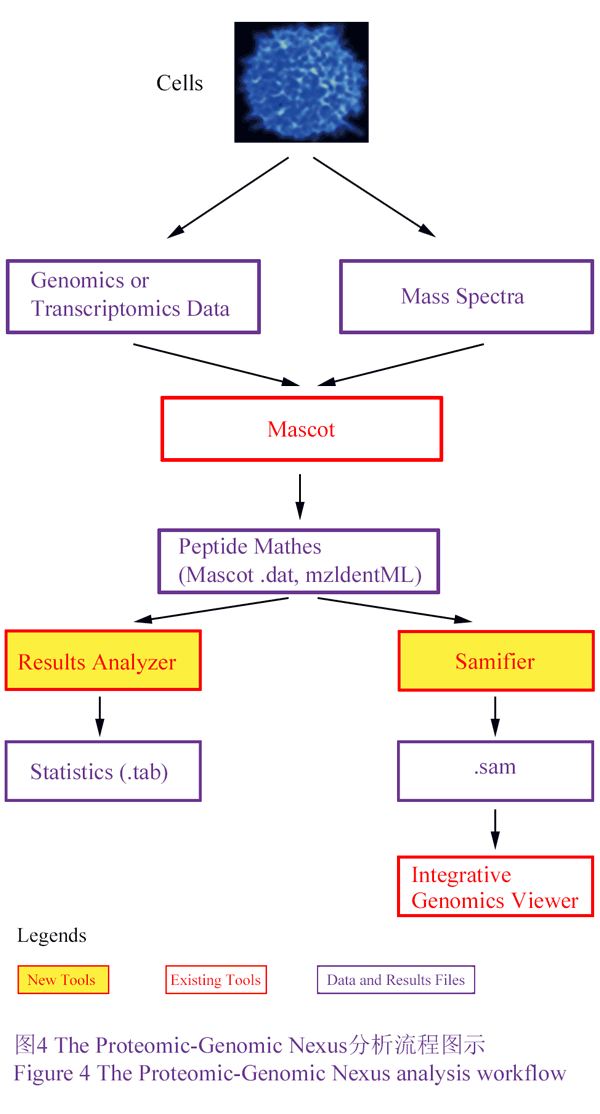
PG Nexus软件包含有两个主要的自主设计的工具:Samifier和Results analyser。Samifier可以将蛋白质质谱转换为SAM格式,这就可以在整合基因组学查看器(integrative genomics viewer, IGV)中同时查看基因组学,转录组学和蛋白质组学的数据。Results analyser报告肽段和蛋白质的数目和类型,并可以报告他们基于自设地订的过滤条件所对应的Mascot得分,跨越外显子直接连接的也被高亮显示,这可被用于验证蛋白质的不同剪切变异体。在分析原核生物的基因组时,PG Nexus多增加了Virtual protein generator和Virtual protein merger两个工具:Virtual protein generator用来产生基于Glimmer基因预测的Mascot序列数据;Virtual protein merger则是通过搜索起始密码子和终止密码子的两侧,重新计算那些匹配到虚拟蛋白质的肽的PG开放阅读框的位置。Nexus软件可以整合入Galaxy项目,这就极大的增强了其使用的方便性。
巩鹏涛负责论文的构思,文献调研,初稿撰写及修改;徐润生负责文献阅读、整理和确认,并对论文提出修改意见;方宣钧博士负责论文写作框架的确定、全文系统修改以及最后的定稿。
Ahmed F. E. 2008, Utility of mass spectrometry for proteome analysis: part I. Conceptual and experimental approaches, Expert Rev Proteomics, 5 (6): 841-864
Ahmed F. E. 2009, Utility of mass spectrometry for proteome analysis: part II. Ion-activation methods, statistics, bioinformatics and annotation, Expert Rev Proteomics, 6 (2): 171-197
Allmer J., Markert C., Stauber E. J., and Hippler M. 2004, A new approach that allows identification of intron-split peptides from mass spectrometric data in genomic databases, FEBS Lett, 562 (1-3): 202-206
Beausoleil S. A., Villen J., Gerber S. A., Rush J., and Gygi S. P. 2006, A probability-based approach for high-throughput protein phosphorylation analysis and site localization, Nat Biotechnol, 24 (10): 1285-1292
Bern M., Cai Y., and Goldberg D. 2007, Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry, Anal Chem, 79 (4): 1393-1400
Castellana N. E., Pham V., Arnott D., Lill J. R., and Bafna V. 2010, Template proteogenomics: sequencing whole proteins using an imperfect database, Mol Cell Proteomics, 9 (6): 1260-1270
Castellana N. E., Payne S. H., Shen Z., Stanke M., Bafna V., and Briggs S. P. 2008, Discovery and revision of Arabidopsis genes by proteogenomics, Proc Natl Acad Sci U S A, 105 (52): 21034-21038
Castellana N. E., Shen Z., He Y., Walley J. W., Cassidy C. J., Briggs S. P., and Bafna V. 2014, An automated proteogenomic method uses mass spectrometry to reveal novel genes in Zea mays, Mol Cell Proteomics, 13 (1): 157-167
Chapman Brett, and Bellgard Matthew. 2014, High-throughput parallel proteogenomics: A bacterial case study, PROTEOMICS, 14 (23-24): 2780-2789
Ferro M., Tardif M., Reguer E., Cahuzac R., Bruley C., Vermat T., Nugues E., Vigouroux M., Vandenbrouck Y., Garin J., and Viari A. 2008, PepLine: a software pipeline for high-throughput direct mapping of tandem mass spectrometry data on genomic sequences, J Proteome Res, 7 (5): 1873-1883
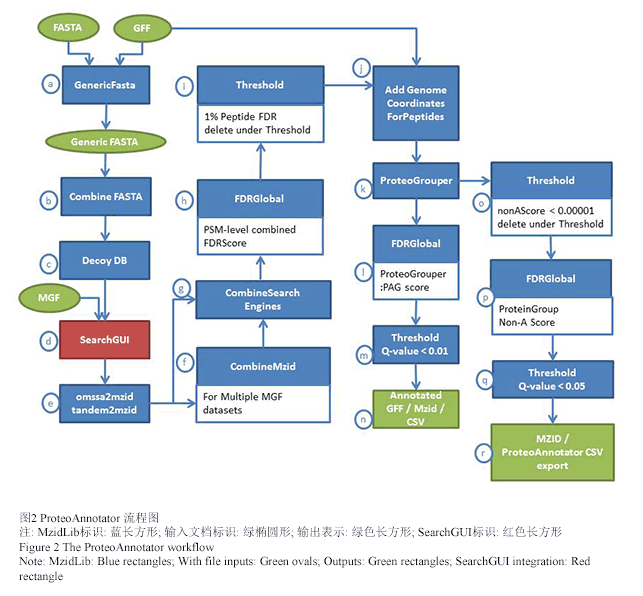
Ghali F., Krishna R., Perkins S., Collins A., Xia D., Wastling J., and Jones A. R. 2014, ProteoAnnotator - Open Source Proteogenomics Annotation Software Supporting PSI Standards, Proteomics, 14 (23-24): 2731-2741
Ghali F., Krishna R., Lukasse P., Martinez-Bartolome S., Reisinger F., Hermjakob H., Vizcaino J. A., and Jones A. R. 2013, Tools (Viewer, Library and Validator) that facilitate use of the peptide and protein identification standard format, termed mzIdentML, Mol Cell Proteomics, 12 (11): 3026-3035
Gong P.T., Xu R.S. Xu and X.J. Fang, Proteogenomics: Progress, Strategy and Problem Genomics and Applied Biology, 2014, Vol.33, No.6, 1169-1180
Jaffe J. D., Berg H. C., and Church G. M. 2004, Proteogenomic mapping as a complementary method to perform genome annotation, Proteomics, 4 (1): 59-77
Jagtap P., Goslinga J., Kooren J. A., McGowan T., Wroblewski M. S., Seymour S. L., and Griffin T. J. 2013, A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies, Proteomics, 13 (8): 1352-1357
Jagtap Pratik Dilip, Johnson James E., Onsongo Getiria, Sadler Fredrik W., Murray Kevin, Wang Yuanbo, Sheynkman Gloria M., Bandhakavi Sricharan, Smith Lloyd M., and Griffin Timothy J. 2014, Flexible and accessible workflows for improved proteogenomic analysis using the Galaxy framework, Journal of Proteome Research, 13 (12): 5898-5908
Jones A. R., Siepen J. A., Hubbard S. J., and Paton N. W. 2009, Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines, Proteomics, 9 (5): 1220-1229
Kim Min-Sik, Pinto Sneha M., Getnet Derese, Nirujogi Raja Sekhar, Manda Srikanth S., Chaerkady Raghothama, Madugundu Anil K., Kelkar Dhanashree S., Isserlin Ruth, Jain Shobhit, Thomas Joji K., Muthusamy Babylakshmi, Leal-Rojas Pamela, Kumar Praveen, Sahasrabuddhe Nandini A., Balakrishnan Lavanya, Advani Jayshree, George Bijesh, Renuse Santosh, Selvan Lakshmi Dhevi N., Patil Arun H., Nanjappa Vishalakshi, Radhakrishnan Aneesha, Prasad Samarjeet, Subbannayya Tejaswini, Raju Rajesh, Kumar Manish, Sreenivasamurthy Sreelakshmi K., Marimuthu Arivusudar, Sathe Gajanan J., Chavan Sandip, Datta Keshava K., Subbannayya Yashwanth, Sahu Apeksha, Yelamanchi Soujanya D., Jayaram Savita, Rajagopalan Pavithra, Sharma Jyoti, Murthy Krishna R., Syed Nazia, Goel Renu, Khan Aafaque A., Ahmad Sartaj, Dey Gourav, Mudgal Keshav, Chatterjee Aditi, Huang Tai-Chung, Zhong Jun, Wu Xinyan, Shaw Patrick G., Freed Donald, Zahari Muhammad S., Mukherjee Kanchan K., Shankar Subramanian, Mahadevan Anita, Lam Henry, Mitchell Christopher J., Shankar Susarla Krishna, Satishchandra Parthasarathy, Schroeder John T., Sirdeshmukh Ravi, Maitra Anirban, Leach Steven D., Drake Charles G., Halushka Marc K., Prasad T. S. Keshava, Hruban Ralph H., Kerr Candace L., Bader Gary D., Iacobuzio-Donahue Christine A., Gowda Harsha, and Pandey Akhilesh. 2014, A draft map of the human proteome, Nature, 509 (7502): 575-581
Kim S., and Pevzner P. A. 2014, MS-GF+ makes progress towards a universal database search tool for proteomics, Nat Commun, 5: 5277
Krug Karsten, Nahnsen Sven, and Macek Boris. 2011, Mass spectrometry at the interface of proteomics and genomics, Molecular BioSystems, 7 (2): 284-291
Kucharova V., and Wiker H. G. 2014, Proteogenomics in microbiology: Taking the right turn at the junction of genomics and proteomics, Proteomics: Kuhring M., and Renard B. Y. 2012, iPiG: integrating peptide spectrum matches into genome browser visualizations, PLoS One, 7 (12): e50246
Kumar D., Yadav A. K., Kadimi P. K., Nagaraj S. H., Grimmond S. M., and Dash D. 2013, Proteogenomic analysis of Bradyrhizobium japonicum USDA110 using GenoSuite, an automated multi-algorithmic pipeline, Mol Cell Proteomics, 12 (11): 3388-3397
Nesvizhskii A. I. 2010, A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics, J Proteomics, 73 (11): 2092-2123
Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., and Horning S. 2009, A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed, Mol Cell Proteomics, 8 (12): 2759-2769
Pang C. N., Tay A. P., Aya C., Twine N. A., Harkness L., Hart-Smith G., Chia S. Z., Chen Z., Deshpande N. P., Kaakoush N. O., Mitchell H. M., Kassem M., and Wilkins M. R. 2014, Tools to covisualize and coanalyze proteomic data with genomes and transcriptomes: validation of genes and alternative mRNA splicing, J Proteome Res, 13 (1): 84-98
Peterson E. S., McCue L. A., Schrimpe-Rutledge A. C., Jensen J. L., Walker H., Kobold M. A., Webb S. R., Payne S. H., Ansong C., Adkins J. N., Cannon W. R., and Webb-Robertson B. J. 2012, VESPA: software to facilitate genomic annotation of prokaryotic organisms through integration of proteomic and transcriptomic data, BMC Genomics, 13: 131
Renuse S., Chaerkady R., and Pandey A. 2011, Proteogenomics, Proteomics, 11 (4): 620-630
Risk B. A., Edwards N. J., and Giddings M. C. 2013a, A peptide-spectrum scoring system based on ion alignment, intensity, and pair probabilities, J Proteome Res, 12 (9): 4240-4247
Risk B. A., Spitzer W. J., and Giddings M. C. 2013b, Peppy: proteogenomic search software, J Proteome Res, 12 (6): 3019-3025
Roos F. F., Jacob R., Grossmann J., Fischer B., Buhmann J. M., Gruissem W., Baginsky S., and Widmayer P. 2007, PepSplice: cache-efficient search algorithms for comprehensive identification of tandem mass spectra, Bioinformatics, 23 (22): 3016-3023
Sanders W. S., Wang N., Bridges S. M., Malone B. M., Dandass Y. S., McCarthy F. M., Nanduri B., Lawrence M. L., and Burgess S. C. 2011, The proteogenomic mapping tool, BMC Bioinformatics, 12: 115
Sevinsky J. R., Cargile B. J., Bunger M. K., Meng F., Yates N. A., Hendrickson R. C., and Stephenson J. L., Jr. 2008, Whole genome searching with shotgun proteomic data: applications for genome annotation, J Proteome Res, 7 (1): 80-88
Shadforth I., Xu W., Crowther D., and Bessant C. 2006, GAPP: a fully automated software for the confident identification of human peptides from tandem mass spectra, J Proteome Res, 5 (10): 2849-2852
Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., and Hunt D. F. 2004, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry, Proc Natl Acad Sci U S A, 101 (26): 9528-9533
Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., and Bafna V. 2005, InsPecT: identification of posttranslationally modified peptides from tandem mass spectra, Anal Chem, 77 (14): 4626-4639
Thelen J. J., and Miernyk J. A. 2012, The proteomic future: where mass spectrometry should be taking us, Biochem J, 444 (2): 169-181
Tovchigrechko A., Venepally P., and Payne S. H. 2014, PGP: parallel prokaryotic proteogenomics pipeline for MPI clusters, high-throughput batch clusters and multicore workstations, Bioinformatics, 30 (10): 1469-1470
Uszkoreit Julian, Plohnke Nicole, Rexroth Sascha, Marcus Katrin, and Eisenacher Martin. 2014, The bacterial proteogenomic pipeline, BMC Genomics, 15 (Suppl 9): S19
Vaudel M., Barsnes H., Berven F. S., Sickmann A., and Martens L. 2011, SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches, Proteomics, 11 (5): 996-999
Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., and Hermjakob H. 2013, The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013, Nucleic Acids Res, 41 (Database issue): D1063-1069
Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. 2014, ProteomeXchange provides globally coordinated proteomics data submission and dissemination, Nat Biotechnol, 32 (3): 223-226
Wenger C. D., and Coon J. J. 2013, A proteomics search algorithm specifically designed for high-resolution tandem mass spectra, J Proteome Res, 12 (3): 1377-1386
Wenger C. D., McAlister G. C., Xia Q., and Coon J. J. 2010, Sub-part-per-million precursor and product mass accuracy for high-throughput proteomics on an electron transfer dissociation-enabled orbitrap mass spectrometer, Mol Cell Proteomics, 9 (5): 754-763
Xu Q., and Lee C. 2003, Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences, Nucleic Acids Res, 31 (19): 5635-5643
Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., Liebler D. C., the Nci Cptac, and National Cancer Institute Clinical Proteomics Tumor Analysis Consortium. 2014, Proteogenomic characterization of human colon and rectal cancer, Nature, 513 (7518): 382-387
Zhou C., Chi H., Wang L. H., Li Y., Wu Y. J., Fu Y., Sun R. X., and He S. M. 2010, Speeding up tandem mass spectrometry-based database searching by longest common prefix, BMC Bioinformatics, 11: 577

 作者
作者  通讯作者
通讯作者